


MLCについて


MLCとは
概要

皮質下嚢胞をもつ大頭型白質脳症(Megalencephalic leukoencephalopathy with subcortical cysts;MLC)は,1995年にオランダのvan der knaap(ファンデルナップ)により報告された白質ジストロフィー※1のひとつです。症状として,大頭症,発達遅延,運動失調,痙縮,軽度認知障害,運動機能障害,てんかんなどが報告されていますが,発症年齢や進行速度は人によってさまざまです。疾患のタイプとして,症状が進行するMLC1,MLC2A,症状が改善あるいは正常化する寛解型のMLC2Bの3つに分類されます※2。MLC1型が本疾患の約76%を占めます。
※1 白質ジストロフィーとは
Leukodystrophy(白質ジストロフィー) は、白質脳症のうち遺伝的欠陥によって引き起こされる疾患であり、病理学的には進行性の症状とミエリン(髄鞘)障害によって特徴づけられます。ミエリンは,脳内の信号を伝達する軸索を覆う構造体であり,グリア細胞のひとつであるオリゴデンドロサイトにより作成されます。ミエリンの存在により脳内の信号伝達が円滑に行うことができますが,白質ジストロフィーでは,ミエリンに障害があるため信号伝達が円滑にいかず,様々な神経機能障害が引き起こされます。
よく似た用語にLeukoencephalopathy(白質脳症)がありますが,これはより広範な定義で,白質のみ,あるいは主に白質に関与する全ての障害のことを言い,遺伝性でない後天的な疾患や,進行性でないものも含まれます。例えば,多発性硬化症や進行性多巣性白質脳症(PML)などは,白質ジストロフィーではない白質脳症です。
※2 MLCのタイプについて
2023年の論文(Passchier et al.,2023)で,これまで臨床症状とMRIからMLCと診断されたが変異遺伝子が不明であった患者から,新しい2つの変異遺伝子GPRC5B,AQP4が発見され,それぞれMLC3,MLC4と命名されました。
MLCのタイプ
| 表現型 | タイプ | 変異遺伝子 | 患者数割合 | |
|---|---|---|---|---|
| 古典型MLC | MLC1 | MLC1 | 常染色体劣性遺伝 | 約76% |
| MLC2A | HEPACAM(GlialCAM) | 常染色体劣性遺伝 | MLC2Bと合わせて約22% | |
| MLC3 | GPRC5B | 常染色体優性遺伝(2023年時点) | 1%未満 | |
| 寛解型MLC | MLC2B | HEPACAM(GlialCAM) | 常染色体優性遺伝 | MLC2Aと合わせて約22% |
| MLC4 | AQP4 | 常染色体劣性遺伝 | 1%未満 | |
| 不明 | 1%未満 | |||
症状
ほとんどの症例で最初に現れる症状は,生後早期の大頭症(頭囲の拡大)です。また,多くの症例で発達の軽度の遅延が見られます。
MLC1,MLC2Aの患者では,運動失調,痙縮,軽度認知障害,運動機能障害,てんかんなどの神経症状が現れます。症状は進行性の経過をたどる場合がほとんどですが、発症年齢や進行速度には人により大きなばらつきがあります。発症後数年で歩く能力を失いほとんどの日常生活に介助が必要となる人もいれば,成人期でも歩くことができ通常の仕事をする人もいます。今のところ,このばらつきの要因を説明できる知見は得られていません。頭部外傷や発熱を契機に症状が悪化する場合があります。
MLC2Bの患者では,一部不器用さや筋緊張の低下が見られますが,運動発達は概ね良好です。てんかんが見られる場合があります。また,自閉症や知的障害を伴う例も報告されています。

病因
MLC1型の原因として、22番染色体の22q13.33という位置に存在するMLC1遺伝子が見つかっています。MLC1遺伝子は、脳および脾臓、白血球などに見られるMLC1タンパク質の設計図となる遺伝子です。MLC1型は、常染色体劣性(潜性)遺伝形式で遺伝します。MLC2A型、および2B型の原因遺伝子として、11番染色体の11q24.2という位置に存在するHEPACAM遺伝子が見つかっています。HEPACAM遺伝子は、脳のグリア細胞や肝臓で見られるGlialCAMタンパク質の設計図となる遺伝子です。MLC2A型は常染色体劣性(潜性)遺伝形式で、MLC2B型は常染色体優性(顕性)遺伝形式で遺伝します(遺伝性疾患プラス)。
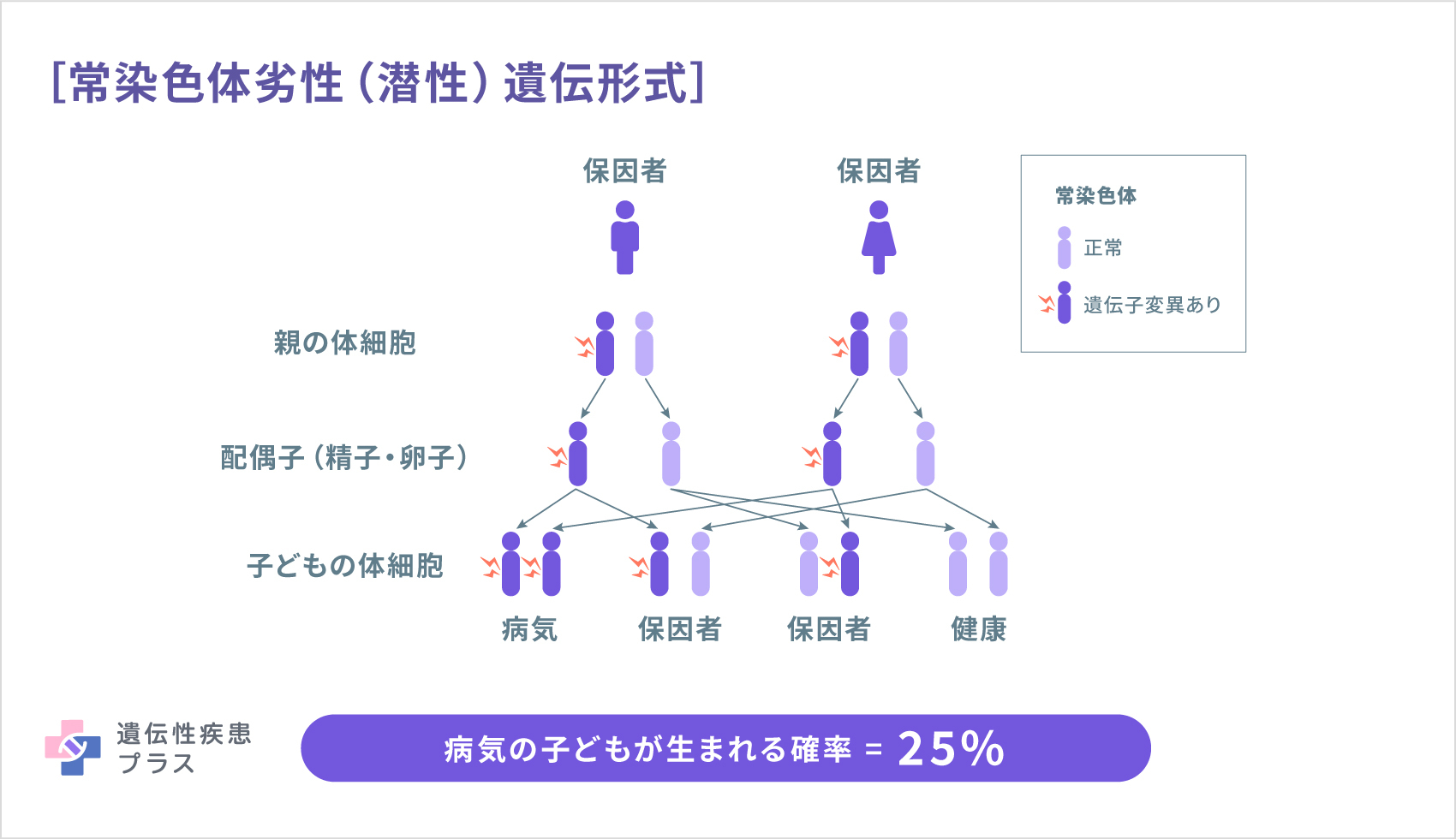
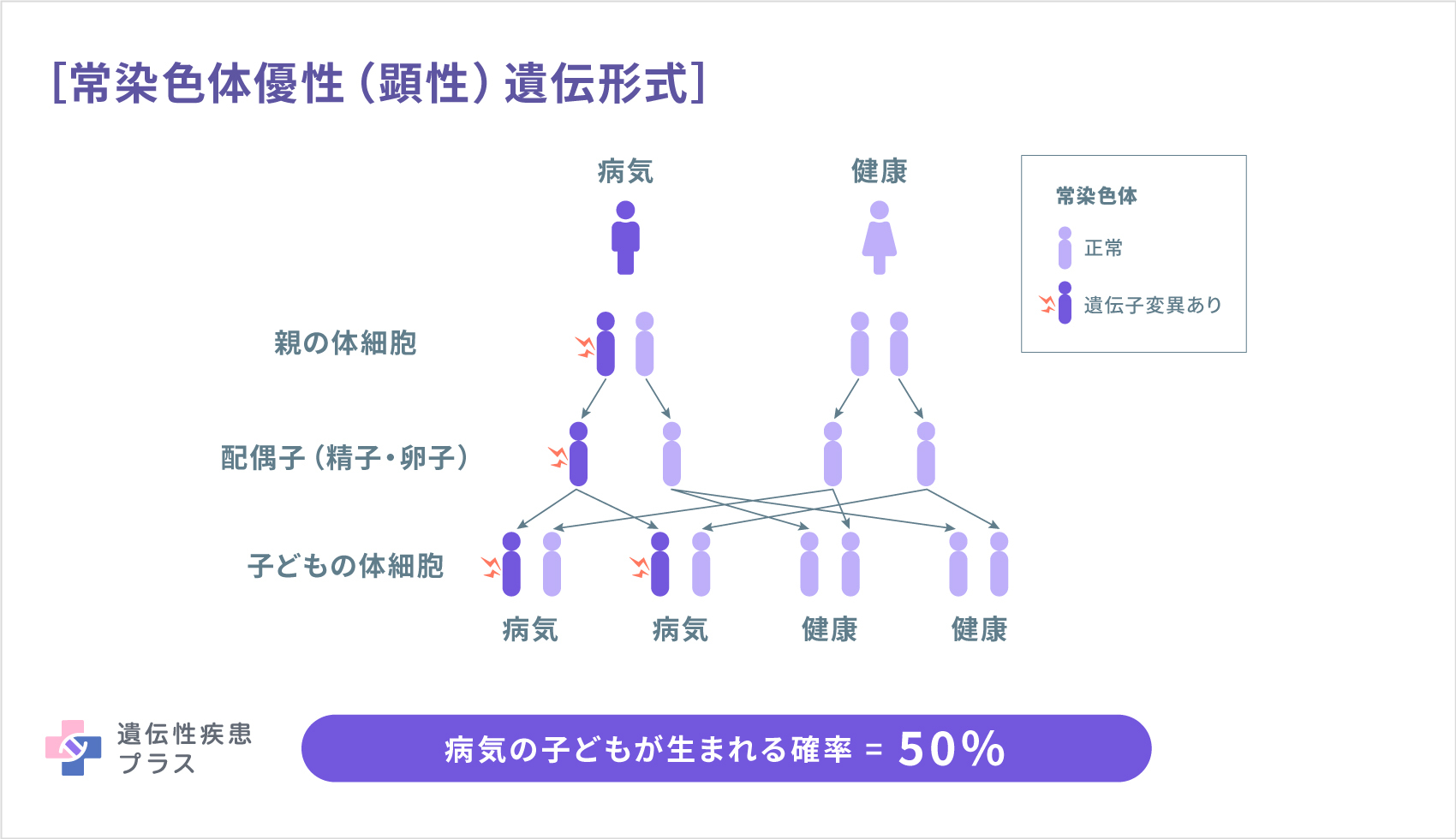
遺伝形式(遺伝性疾患プラスより転載)
MLC1タンパク質,GlialCAMタンパク質はいずれもグリア細胞のひとつであるアストロサイト(星状細胞)に主に存在し,グリア細胞のイオンや水の恒常性を保つ役割を持つ別のタンパク質(ClC-2チャネル,Na+/K+-ATPアーゼ,Ca2+-ATPアーゼ,V-ATPアーゼ,VRACチャネル,Cx43,TRPV4,Kir4.1,AQP4,など)の活性を制御する役割があると考えられています(Bosch et al.,2021)。
遺伝子変異によりMLC1あるいはGlialCAMがうまく働かない,あるいは,いるべき部位に生成されない(誤局在;mislocalization)ことで,上記イオンチャネルの活性化制御など本来の役割を果たせず,グリア細胞のイオン・水のバランスが崩れます。その結果(具体的なメカニズムは不明ですが),細胞体積の膨張や,同じグリア細胞であるオリゴデンドロサイトにより生成されるミエリンの空胞化(Vacuolization)が引き起こされます。ミエリンは,脳内の信号を伝達する軸索を覆う構造体であり,信号伝達速度を飛躍的に向上させる役割がありますが,その機能が空胞化により障害されることで,様々な神経機能障害が症状として現れると考えられています。
上記のグリア細胞の恒常性を保つ働きは定常的に行われていることですが,頭部外傷や発熱時にはより活発化するため,その制御がうまくいかない場合には定常時よりもさらに大きな影響を受けます。つまり,頭部外傷や発熱時に階段状に症状が進む場合があるというMLCの特徴は,この理由により説明できると考えられています。
なお,MLCの特徴のひとつである皮質下嚢胞の発生メカニズムや嚢胞による臨床症状への影響などは,明らかにされていません。
診断方法
MRIによる画像診断と臨床的兆候により診断されます。MRI画像は,特徴的なびまん性の大脳白質信号異常と,皮質下嚢胞の存在を示します。最終的には,遺伝子検査によるMLC1およびHEPACAM遺伝子の変異を確認することで確定されますが,どちらの変異も確認されず臨床症状とMRIにより診断される例も稀にあります。
治療

現在,確立された治療法はありません。発作を抑えるための抗てんかん薬など,発生した症状に対する支持的療法に留まります。
ただし,現在も日本及び諸外国で研究が続けられており,病態解明や治療法に関する知見は増えてきています。人間を対象とした臨床試験はまだ行われた例がありませんが,マウスやiPS細胞などを用いた病理モデルで遺伝子治療や分子標的薬などの既存薬の有効性を検証する研究が行われています。
・遺伝子治療
疾患の根本原因であるタンパク質(MLCで言うとMLC1あるいはGlialCAM)を修正するアプローチです。手法としてAAV(アデノウィルス随伴ベクター)を用いた遺伝子導入やLV(レンチウィルスベクター)を用いた造血幹細胞移植などが主流です。疾患の根本原因を修正するアプローチなので,MLC1やGlialCAMがほかのタンパク質に与える影響など,下流の複雑なメカニズムが完全に明らかでなくても治療法を確立することが可能と考えられます。Sánchez et al.(2020)では,MLC1のノックアウトマウスを対象とした遺伝子治療が行われており,AAVベクターによるアストロサイトをターゲットとした遺伝子導入を行い,ミエリン空胞化が有意に減少したことが示されています。しかし,マウスモデルが,人間のMLC患者の表現型を完全に再現できているわけではないことなど,人間を対象とした治療への展開においては課題が残されています。
・既存薬による治療
MLC1あるいはGlialCAMは,グリア細胞のイオン・水の恒常性に関与する他のタンパク質の活性を制御することが示されています。従って,遺伝子変異により失われたこれらの役割を薬剤により代替する治療アプローチが考えられます。例えば,Lanciotti et al.(2020),XabierElorza-Vidal et al.(2018)では,MLC1タンパクが正常に機能しない場合には,タンパク質の情報伝達経路であるシグナル分子「ERKキナーゼ」を経由して,前述のCx43やVRACなどのタンパク質が過剰に活性化されることが示されており,ERKキナーゼの活性化を阻害する薬剤の有効性が示唆されています。また,Lanciotti et al.(2016)では,MLC1変異が,アストロサイトなどの細胞増殖に関わるシグナル伝達分子「上皮成長因子受容体(EGFR)」の制御機構がうまく働かないため,細胞増殖が過剰になることが示されており,EGFRチロシンキナーゼ活性を阻害する薬剤の有効性が示唆されています。上記のシグナル分子に働きかける薬剤は「分子標的薬」と呼ばれており,がんの治療用として承認されている薬も多くあるため,遺伝子治療よりも早く治療法を確立できる可能性があります(つまり部分的にがんと同じメカニズムということになりますが,MLC患者において脳腫瘍が増加したという報告は今のところありません)。
ただし,複雑なMLCの病態の全体像が完全に明らかにされているわけではないので,特定の分子を標的とした薬剤で,大局的な効果が得られるかどうかはわかりません。
MLC1,2Aは基本的には進行性の疾患であり,治療を行わない限り運動機能・認知機能は速度の個人差はあれどほぼ例外なく退行の一途をたどると考えられています。しかし,治療研究において,ミエリン空胞化がかなり進んだ15か月齢のマウスにおいても遺伝子治療によりミエリン空胞化が正常化したこと(Sánchez et al.,2020)から,MLCは顕著な神経変性(細胞死など)はなく臨床表現型は可逆的である可能性があることが示唆されています。また,人間の表現型においても,Van der knaap(2016)において, MLCを含むミエリン空胞化を伴う白質ジストロフィーで可逆性を示唆する以下のような知見がレビューされています。
・寛解型であるMLC2Bでは,生後1~2年で一旦ミエリンの空胞化とみられるMRI白質信号異常が確認された後,その異常が正常化するとともに運動機能も正常化あるいは改善する(Hamilton et al., 2018)。
・GJB1遺伝子変異によるX連鎖シャルコー・マリー・トゥース病では,ミエリン空胞化とみられる一過性のMRI白質信号異常が発症する可能性がある(Depienne et al.2013,Paulson et al.,2002)。
・脳幹および脊髄の障害と乳酸上昇を伴う白質脳症(LTBL)では、乳児期にMRIで重度の白質脳症と拡散制限を伴う神経学的悪化を特徴とし、ミエリン微小空胞化を示唆しているが、その後,臨床およびMRIが改善され、ほとんどの異常が消失する症例がある(Steenweg et al.2012)。
・新生児メープルシロップ尿症は、新生児のミエリンを含む領域の急性ミエリン微小空胞化を特徴とし、異常は適切な治療で回復可能である(Jan et al.,2003)。
従って,ミエリン空胞化自体とそれに起因する臨床症状は可逆的であり,ある程度症状が進行しても広い治療範囲が残っている可能性があります。ただし,自閉症や認知機能低下などの他の表現型は、ミエリン空胞化の影響であるかどうか不明なので,同じように可逆的であるかどうかはわかっていません。
患者数
極めて稀な病気であるため,患者数について明確な報告はありませんが,日本では20人程度と言われています。過去の症例報告では,最も大規模な研究(Hamilton et al.,2018)で,世界中の約240人の症例が紹介されています。
分類
一般に,白質ジストロフィー(Leukodystrophy)あるいは遺伝性白質脳症という大きなカテゴリーに分類されていますが,細分類として「星状細胞(アストロサイト)の疾患」に分類されます。同じカテゴリーに属する疾患として,
・白質消失病(Vanishing white matter disease;VWM)
・エカルディ・グティエール症候群(Aicardi Goutieres syndrome)
・ClC-2チャネル関連疾患
などが挙げられています(Lanciotti et al., 2013,van der Knaap et al., 2017)。同じカテゴリーに属する疾患の研究成果が,MLCに対しても反映できる可能性があります。
別名
最近はほぼ「Megalencephalic leukoencephalopathy with subcortical cysts; MLC」で統一されていますが,MLCが発見された当初の論文などでは,以下の表記が用いられている例もあります。
・Van der Knaap Syndrome(ファンデルナップ病,ファンデルナップ症候群)
・Vacuolating Megalencephalic Leukoencephalopathy with Subcortical Cysts
・LVM
・Leukoencephalopathy with swelling
・Vacuolating leukoencephalopathy(VL)
MLCについて説明したウェブページ
研究グループ
バルセロナ大学(スペイン)
Raul Estevezを中心とした研究グループです。2000年ごろから現在まで,MLCの病態解明に関する数多くの論文を発表しています。Sanchez et al.(2020)では,遺伝子治療の検討が行われています。MLC1に罹患したマウスに対し,正常な遺伝子を導入することで,症状の改善を達成したことが報告されています。
バルセロナ大学神経科学研究所(The Institute of Neurosciences)ホームページ
イタリア国立衛生研究所(Istituto Superiore di Sanita; ISS; イタリア)
Elena Ambrosiniを中心とした研究グループです。バルセロナ大学同様に,MLCに関する数多くの論文を発表しています。Lanciotti et al.(2020)では,患者由来のiPS細胞から分化した星状細胞を用いた研究が現在進行中であることが示されています。ヒトiPS細胞を用いることで,これまでより詳細な病態解明が期待されます。また,さまざまな薬の効果をin vitro(試験管内)で試すことができるため,既に承認されている薬の有効性が明らかになる可能性もあります。
アムステルダム自由大学(VU大学; オランダ)
MLCを最初に発見したvan der Knaapが所属する大学です。古典的MLCと呼ばれる「MLC1型」を発見したのち,GliamCAM変異による「MLC2A型」,「MLC2B型」を発見(Lopez-Hernandez et al.,2011)するなど,MLCのパイオニアです。Hamilton et al.(2018)では,世界中242人の患者データに基づく分析が行われており,これまでで最大規模の症例報告です。
van der Knaapは同じアストロサイト疾患である白質消失病(VWM)の発見者でもあり,VWMの研究にも注力しています。最近では,VWMの治療法に関する研究(Dooves et al., 2017,Dooves et al., 2019),iPS細胞を用いた病態解明(Leferink et al., 2019)が発表されています。
アムステルダム自由大学UMC(University Medical Centers)ホームページ
慶応義塾大学(日本)
慶応義塾大学の田中謙二先生らにより開発された多機能遺伝子改変システム(FASTシステム)を用いたMLCの病態解明(Tanaka et al.,2010,Sugio et al.,2017,Kikuchihara et al.,2018)が発表されています。
また,2023年よりMLCのAAVベクターによる遺伝子治療の研究に着手していただいています。
【新着情報】日本初 MLCの遺伝子治療に予算がつきました。(2023.04.15)
【新着情報】慶応義塾大学、田中先生、生田さんにお時間をいただきました。(2023.09.16)
【新着情報】MLC遺伝子治療開発にAMED大型予算がつきました。(2024.03.11)
東京女子医科大学(日本)
当会顧問の山本俊至先生が所属する大学です。MLCの症例報告(Shimada et al., 2014,Masuda et al.,2015,Yamamoto-Shimojima et al.,2020)が発表されています。
北京大学
MLCの症例報告(Wang et al.,2011,Cao et al.,2016)や,病態解明(Xie et al.,2012,Shi et al.,2019)が発表されています。また,類似疾患である白質消失病(VWM)に対する患者iPSC由来のアストロサイトを用いた病態解明(Zhou et al.,2019, Deng et al.(2022))も発表されています。
韓国脳研究所(Korea Brain Research Institute;KBRI;韓国)
MLCの病態解明に関する論文(Hwang et al.,2019,Hwang et al.,2021)が発表されています。
その他にも以下のような症例報告が発表されています。
・Tsujino et al.(2003)(国立精神神経医療研究センターほか; 日本)
・Dai et al.(2017)(中南大学; 中国)
・Choi et al.(2017)(ソウル国立大学ほか; 韓国)
